Gyógyíthatatlannak gondolt betegségekre hozhat megoldást a génterápiák új generációja
„Hogy vezethetett egy ennyire szép dolog egy ilyen szörnyűséghez?”
- ez a nem is annyira költői kérdés Sid Mukherjee lebilincselő The Gene: An Intimate History című könyvében hangzik el a génterápiák évtizedes történetének egyik legtragikusabb balesete kapcsán Paul Gelsinger szájából. Paul, annak a tinédzser fiúnak, Jesse Gelsingernek az édesapja, akinek a tizennyolc évvel ezelőtti halála közel másfél évtizedre takaréklángra tette az akkoriban még nem kiforrott, ellenben jelentősen túlhájpolt génterápiás beavatkozásokat, hogy aztán a megfelelő tanulságok levonása után azok napjaink legígéretesebb gyógykezeléseiként tűnjenek újból fel. És ez a reneszánsz épp annak köszönhetően lett lehetséges, hogy mostanra pontosa(bba)n látjuk, az alapelvében elegánsan egyszerűnek tűnő génterápiás beavatkozások milyen nem triviális veszélyeket rejtenek.
A génterápia alapgondolata, vagyis az az elgondolást, hogy bizonyos mutációk estén a hibás, vagy hiányzó fehérjét kódoló gén működőképes verzióját vírusok segítségével lehetne bejuttatni a beteg sejtjeibe, már bő fél évszázados múltra tekint vissza. Az 1960-as évek végén a nyulakat megfertőző Shope papillomavírussal fogallakozó amerikai Stanfield Rogers-ben ötlött fel először, hogy a Shope-t és más vírusokat terápiás célra lehetne használni. A humán HPV-vel rokon Shope vírus a nyulakon kemény, szemölcs-szerű keratinkinövéseket hoz létre (néha egészen extrém és ijesztő formában), miközben amolyan mellékhatásként a fertőzött sejtek túlexpresszálják az arginin nevű aminosav bontásáért felelős argináz enzimet, így ezeknek a sejteknek kifejezetten alacsony arginin tartalmuk van. Rogers feltételezte, hogy a vírus valamiképpen tartalmazza az argináz enzimet, így amikor pár évvel később egy olyan német testvérpárba botlott, akiknek a szervezete túl sok arginint hozott létre, kapva-kapott az alkalmon, hogy tesztelje az ötletét.
A hiperargininémia nem kellemes betegség: egyre durvuló epilepsziás rohamok, fokozódó izommerevség és koordinációs zavarok jellemzik, gyakran szellemi retardációval jár. Így természetesen az érintett családok szinte bármilyen reménysugárba hajlandók belekapaszkodni, ami terápiás haszonnal kecsegtet, nem meglepő tehát, hogy Rogers is viszonylag könnyen rábírta a német családot, hogy legyenek alanyai az úttörőnek számító vírusterápiás kísérletben. Sajnos azonban a kísérlet nem járt sikerrel, a víruskezelés nem hozta meg a kívánt hatást.
Ma már tudjuk, hogy a Shope vírus egyáltalán nem is tartalmazza az argináz enzimet, és emiatt is az egész kísérlet kifejezetten problémás volt, nem véletlen, hogy Rogerst később hivatalosan is megrótták a beavatkozásért, és többet nem kísérelt meg hasonlót.
Ugyanakkor ez a korai, átgondolatlan kísérlet mégis elegendő volt, hogy kiengedje a palackból a szellemet, és forradalmasítsa a szakma gondolkozását. A következő években a molekuláris biológia forradalma lehetővé tette nemcsak tetszőleges DNS-darabok összeragasztását (ún. rekombináns DNS létrehozását), de egyre több ismeret gyűlt össze a különböző vírusokról is, amelyeket potenciálisan ezeknek a DNS daraboknak a bejuttatására lehetne használni. Így az 1990-es évek közepére minden adott volt, hogy a génterápia kipróbálásra kerüljön mind Európában, mind az Egyesült Államokban.
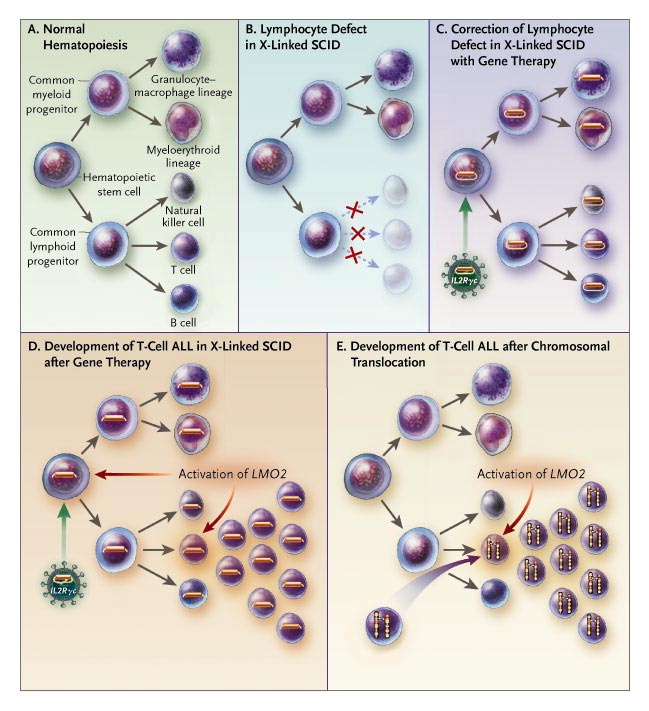
Az európai kísérletek központi alakja a francia Alain Fischer lett, aki az ún. súlyos, kombinált immundefektus (SCID - Severe Combined Immunodeficiency) betegségben szenvedő gyerekek gyógyítására akart génterápiás beavatkozást kifejleszteni. A SCID gyerekek immunrendszere annyira gyenge, hogy a legenyhébb fertőzés is halálos lehet, ennek megfelelően életüket a szó szoros értelemben egy buborékban kell töltsék és szinte senkivel nem érintkezhetnek. Alapesetben egy csontvelő-átültetéssel a SCID számos formáját kezelni lehet, de vannak súlyos esetek, ahol nincs megfelelő donor, és ilyenkor a génterápia lehet a legjobb opció.

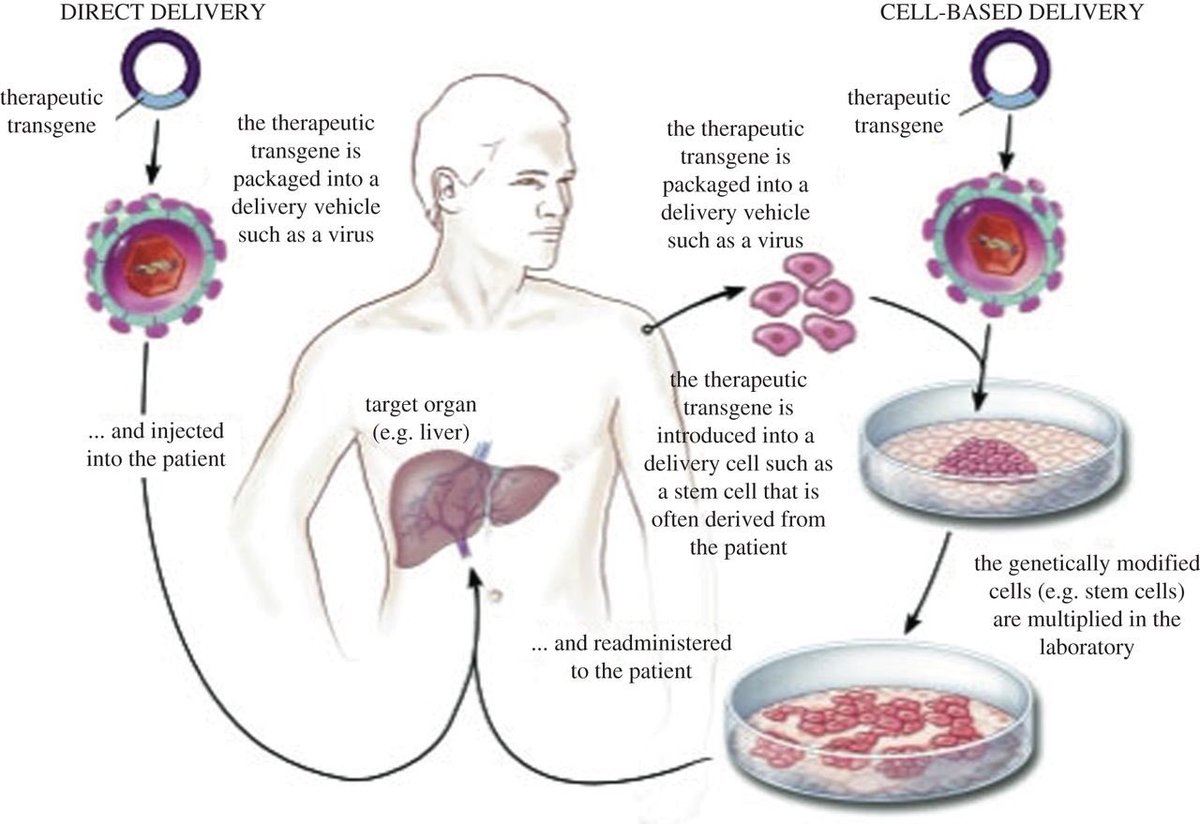
Fischer csoportja a SCID egy olyan típusát tanulmányozta (SCID-X1), ahol az immundefektust okozó mutáció az X-kromoszóma egyik jól jellemzett génjében van, és ennek a génnek a működő variánsát vitték be vérképző őssejtekbe. (Mivel maga a géntranszfer a páciens testén kívül, a laborban történt, és a kezelt őssejteket ültették vissza a betegbe, ezt a kezelési formát ex vivo terápiának hívjuk, megkülönböztetve a páciens testében történő in vivo beavatkozásoktól.) A beavatkozás sikeresnek bizonyult, és a húsz kezelt beteg közül tizenkilenc még mindig életben van.
A Fischer-csoport kísérleteivel szinte párhuzamosan, Philadelphiában, a génterápia-kutatás fellegvárának számító University of Pennsylvania gyerekkórházában (CHOP) egy genetikai hátterű anyagcserebetegség génterápiás gyógyításának tesztelését kezdték el. Az ornitin-transzkarbamiláz (OTC) nevű, májban képződő enzim hiányában a sejtekben a fehérjék lebontása nem megfelelően zajlik le, ammónia halmozódik fel a testben, ami először az ereket teszi tönkre, majd a vér-agy gáton átjutva lassan az idegsejteket is elpusztítja. Az OTC-hiányban szenvedő betegek ritkán élik túl a gyerekkort, és még az extrém, fehérjementes diéta is csak lassítani tudja a folyamatot, leállítani nem, és bármilyen kilengés akár halálos kimenetelű lehet.
A betegség enyhébb tüneteit mutató Jesse Gelsinger például egy mogyoróvajas szendvics elfogyasztása után hosszabb időre kómába került,
nem csoda, hogy a család nagy reményeket fűzött a philadelphiai génterápiához. A génterápia logikussága és szépsége az akkor 17 éves Jesse-t és édesapját is lenyűgözte, így kifejezetten várakozva feküdt be a kórházba. Sajnos azonban már sosem jöhetett ki: a génterápiánál használt vírus különösen erős immunreakciót indukált benne, és az ezzel járó sejtkárosodást az OTC hiányában nem tudta a szervezet feldolgozni. Jesse szervezi fokozatosan felmondták a szolgálatot és néhány nappal később, immár agyhalott állapotban, apja kérésére kapcsolták le a lélegeztetőgépről. Az ügyből hatalmas botrány lett, számos hiányosságot tárt fel egy belső vizsgálat, és a terápiát kiötlő orvost, James Wilsont évekre eltiltották a hasonló munkától.
Hirtelen megsokasodtak a kérdőjelek a korábban sztárolt génterápia körül, és az sem segített, hogy pár évvel később, 2001-ben Alain Fischer arról számolt be, hogy az egyik génterápián átesett betegük leukémiás lett. A betegség oka ugyanis az volt, amit máig a génterápia Achilles-ínjának tartunk: hogy a vírus által bejuttatott DNS darab viszonylag véletlenszerűen ilesztődik be a gazdasejt genomjába, és ritkán, de előfordulhat, hogy emiatt rosszindulatú sejtosztódások indulnak el. Ezután a génterápia kapcsán szinte mindenki csak a homlokát ráncolta.
Az alapgondolat azonban nem tűnt el, hiszen, ahogy azt Paul Gelsinger is megfogalmazta, az „túl szép volt” ahhoz, hogy veszni hagyják. De a most már jobban ismert veszélyek ismeretében sok helyen leállították a tervezett génterápiás beavatkozásokat, és hosszas újratervezési és fejlesztési fázis következett.
Ennek az eredményei kezdenek mostanában beérni, és ennek köszönhető, hogy az elmúlt hónapok során egymást érték a sikeres génterápiákról szóló hírek.
Talán az első a sorban egy adrenoleukodisztrófia (ALD) nevű idegrendszeri megbetegedés ellen kidolgozott eljárás volt. Ez a főleg fiúkat érintő betegség az idegsejteket védő mielinhüvely pusztulásával jár. A négy és tíz év között jelentkező első tünetek után felgyorsul a betegség, és az érintettek gyakran megvakulnak és megsüketülnek, epilepsziás rohamjaik lesznek, elveszítik izmaik felett a kontrollt, majd teljes demencia jöhet, vagy a halál.
Az alig egy hónapja közzétett eredmények szerint az ALD ellen kifejlesztett beavatkozás a Fischerék által használt ex vivo logikát követve juttatja be a működőképes gént egy vérképző őssejtbe, majd ezeket ültették vissza. A sejtek közül néhány eljutott az agyba, és ott gliasejtté alakult; de ennek a pár sejtnek a működése - úgy tűnik - elegendő a szimptomák visszaszorítására - a kezelt 17 gyerek közül 15 közel két évvel a beavatkozás után sem mutatja a tüneteket.

Az ex vivo megközelítés azonban nem mindig alkalmazható: ha olyan gént kell kijavítani, amelynek a terméke nem kiválasztódik, hanem a sejten belül kell fontos szerepet eljátszon, akkor csak az in vivo beavatkozás marad opció.
Még az ALD-nél is fiatalabb korosztályt érint a spinális izomdisztófia (SMA1), amelynek eredményeként az alig néhány hónapos gyerekek fokozatosan lebénulnak. Ebben az esetben a gerincvelői motorneuronok életben maradásáért felelős SMN1 gén hibája áll a szomorú tünetegyüttes mögött. Az SMN1 fehérjére azonban nem akármilyen sejtben, hanem konkrétan a neuronokban van szükség, ami pár évvel ezelőtt még igen nehéz probléma elé állította az orvosokat.
Az idegrendszer ugyanis (persze nem véletlenül) különösen védett része a testünknek: a mechanikai védelmet biztosító hártyákon és agy-gerincvelői folyadékon kívül a szervezet az agyat és a gerincvelőt a fertőzésektől és különböző mérgektől egy alig-alig átjárható szűrőrendszer, a vér-agygát segítségével védi. A génterápiás vírusvektorok első generációja nem is nagyon tudott ezen átjutni, így a korai próbálkozások során a módosított vírusokat közvetlenül az agyba injektálták.
A sors különös fintora, hogy az OCT-fiaskó után a klinikai kísérletektől öt évre etiltott James Wilson csoportja talált erre a problémára megoldást: az eltiltása alatt alapkutatáshoz forduló Wilson ugyanis egy olyan új adeno-asszociált vírust (AAV9) fedezett fel, ami képes a vér-agygáton átjutni és hatékonyan fertőzi meg az idegsejteket is.
Az AAV9-alapú vektorok segítségével elég volt egyetlen egy kezelés, és elegendő SMN fehérje termelődött a gyerekek szervezetében, hogy a tünetek egyáltalán ne alakuljanak ki. (Mind az ALD, mind a SMA1 esetében a tünetek megjelenésekor tulajdonképpen már késő, épp ezért csak megfelelő genetikai szűréssel kerülhet sor időben génterápiás beavatkozásra is.)

Az említett betegségek mellett már szinte eltörpül a „csak” vaksággal járó örökletes retina-sorvadás egyik fajtája, amely esetében szintén az elmúlt hetekben kapott zöld fényt az amerikai Élelmiszer- és Gyógyszerbiztonsági Hatóságtól (FDA) egy génterápiás gyógymód forgalmazása. A szembe injektált beavatkozás eredményeként egy évvel később a 20 kezelt személyből 13 egészen halvány (kb. 1 lux erősségű) fényt is képes volt érzékelni.
Míg az előző beavatkozások tulajdonképpen részletesen megtervezett klinikai vizsgálatok eredményeként történtek, van, amikor az orvosok ezek hiányában kénytelenek cselekedni. A 7 éves Hasszán és családja sikeresen menekült el a háború sújtotta Szíriából, és jutott el Németországba, ám ott az orvosok ugyanúgy tehetetlennek tűntek a kisfiú különleges betegségével szemben, mint szülőhazájában.
Alig egy héttel születése után furcsa seb jelent meg az újszülött hátán, ami alapján már a szír orvosok diagnosztizálták az epidermolysis bullosa (EB) nevű betegséget. Az EB-betegek bőréből hiányzik egy kulcsfontosságú fehérje-komplex, ami rugalmasan tartja a szövetet, így az könnyen töredezik, és leválik, sebet hagyva maga után. Hasszán esetében a LAMB3 gén működésképtelensége a tünetek különösen agresszív együttesét hozta létre - végül már testének közel 80%-áról hiányzott a bőr, folyamatos fertőzéseknek és elképzelhetetlen fájdalmaknak téve ki a gyereket. A helyzettel a német orvosok sem tudtak kezdeni semmit, és már azon gondolkoztak, hogy miképp lehetne a kis beteg utolsó napjait megkönnyíteni, amikor egy modenai csoport révén egy különleges lehetőség merült fel. Az EB egy enyhébb formáját már korábban sikeresen kezelő olasz kutatók Hasszán maradék bőrének egy kis darabját izolálva elkezdték az abban levő sejteket lombikokban növeszteni, majd egy génterápiás vírusvektorral „megfertőzve” őket működőképes LAMB3-t juttattak beléjük.

Bőrünk, akárcsak a vér, egyike a hétköznapi életben is folyamatosan megújuló szöveteinknek. A benne levő őssejteknek köszönhetően durván havonta lecsérlődik a bőr, és amíg maguk az őssejtek nem sérülnek meg, vagy pusztulnak el, ez a megújulás folyamatos. A Hasszánból izolált bőrben levő őssejtek, miután beépítették a génterápiával bevitt gént, osztódni kezdtek, és előbb mesterséges felületeken hoztak létre bőrdarabokat, amelyeket később a beteg testére ültettek át. Ezzel a módszerrel közel egy hónap alatt szinte a teljes bőrt sikerült újraépíteni a kisfiún, akit mostanra haza is engedtek a kórházból. Az új bőr egyáltalán nem sérül könnyen, és ugyanolyan jól tapad a testhez, mint egy egészséges emberé.
Természetesen az EB esetében alkalmazott ex vivo megközelítésnek ugyanaz a potenciális problémája, ami Alain Fischerék esetében is annyi fejfájást okozott: a beépülő DNS darabok véletlenszerűen bombázzák a genomot, és mindig van egy bizonyos esélye annak, hogy egy ilyen integráció valami káros folyamatot indukál. A Hasszán bőréből izolált biopsziákban ennek nyoma sincs, de csak úgy lehetne minden génterápia estében biztosra menni, ha véletlenül integrálódó szekvenciák helyett adott helyen lennénk képesek szerkeszteni a genomot.
Ezek az újgenerációs módszerek már régóta foglalkoztatják a kutatók és orvosok fantáziáját, és úgy tűnik, hogy lassan elérkezünk oda, hogy elkezdődnek a komolyabb klinikai vizsgálatok is.
A napokban derült ugyanis ki, hogy megtörtént az első in vivo genomszerkesztéses beavatkozás az Egyesült Államokban. Ennek az alanya egy másik különleges anyagcsere-betegségben, ún. Hunter-szindrómában szenvedő férfi, Brian Madeux volt.
A Hunter-szindrómás betegek (elsősorban férfiak, hiszen ez is egy X-kromoszómához kötött betegség) esetében egyes cukrok lebontása nem történik meg, és ezek felhalmozódása vezet a sejtek, illetve szövetek működésképtelenségéhez. Létezik egyfajta intravénásan adott enzim-szupplementációs terápia, de az nem jelent végleges gyógyulást, ezért is vonzó erre a betegségre is a génterápia.
A Madeux esetében alkalmazott beavatkozás ugyan valóban az első in vivo végzett emberi genomszerkesztés, de a teljes igazsághoz hozzátartozik, hogy ez azért még nem az a génszerkesztés, amit mindenki vár, hiszen nem a rossz gén kijavítására vállalkoztak a kutatók.
Tulajdonképpen a genomszerkesztéssel csak azt próbálják elérni, hogy a működőképes gén ne a genom egy random helyére illesztődjön be, hanem egy jól ismert pontjára, az albumin fehérjét kódoló gén helyére. Ehhez ezen a ponton elhasítják a genomot (nem az újonnan gyakran használt CRISPR rendszert vetik be, hanem a „genomszerkesztő” molekuláris eszközök egy korábbi generációját, cink-ujj fehérjéket (ZFN)), és a májsejtek DNS-javítási mechanizmusát igyekeznek becsapni, hogy a hasított DNS összekapcsolása közben beépítse az I2S gén szekvenciáját. Ez csak a májsejtek kis százalékában történik majd meg (várhatóan), de ez elegendő lesz ahhoz, hogy az enzim innentől kezdve folyamatosan jelen legyen a szervezetben.
Fogas kérdés lesz ezeknek a hosszú előkészületet igénylő beavatkozásoknak az ára. Ha a nyáron bejelentett génterápiás rákgyógyszerekből indulunk ki, itt is sok százezer dollár lesz a végösszeg. Ami borzasztóan sok. Ugyanakkor látható, hogy sok esetben egy egyszeri beavatkozás után már nem lesz további kezelésre szükség, ami mégis költséghatékonnyá teheti a génterápiát. Hiszen egy Hunter-szindrómás beteg esetében az eddig alkalmazott enzim-szupplementáció is évi 100-400 000 dollárba került. És hát abban is bízni lehet, hogy ahogy lassan rutinbeavatkozássá válnak ezek a beavatkozások, az árak meredeken csökkenni fognak, így csak idő kérdése, hogy kvázi rutinalkalmazássá váljanak.
(Forrás: Science, Nature, The Atlantic, The New York Times és megint Science. A fedőkép forrása yourgenome.org.)
Elkezdődött a rákellenes génterápiák kora
A Novartis Kymriah nevű új terápiája a rákellenes küzdelem egy új korszakát nyithatja meg.

Genetikailag Módosított Emberek
A legújabb genomszerkesztési technológiák számos genetikai betegség gyógyításának ígéretét hordozzák. De sokan aggódnak, hogy pontos kockázataiknak ismerete mellett még túl korai lehet emberi alkalmazásuk.